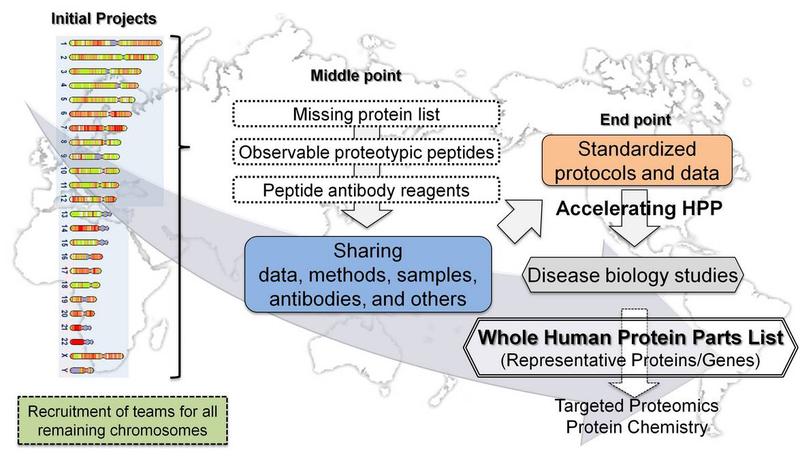
To map the human protein subset or parts list coded by genes on each chromosome.
- Starting Point: Missing Proteins (>30%) Mapping
- End Point: Complete list of all proteins coded by genes in all human chromosomes.
- The mapping will be complemented by studies of cellular and organ expression, subcellular distribution, functions, PTM and interactions, under specific physiological settings, and alterations of expression and isoforms associated with pathophysiology.
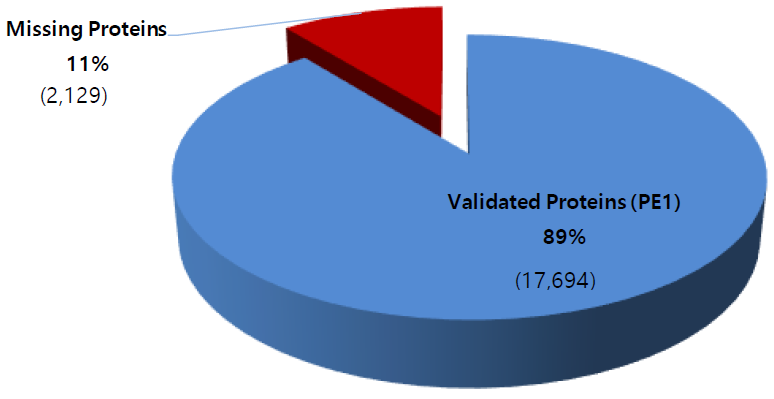
Update on the missing proteins (as of Jan. 11, 2019)
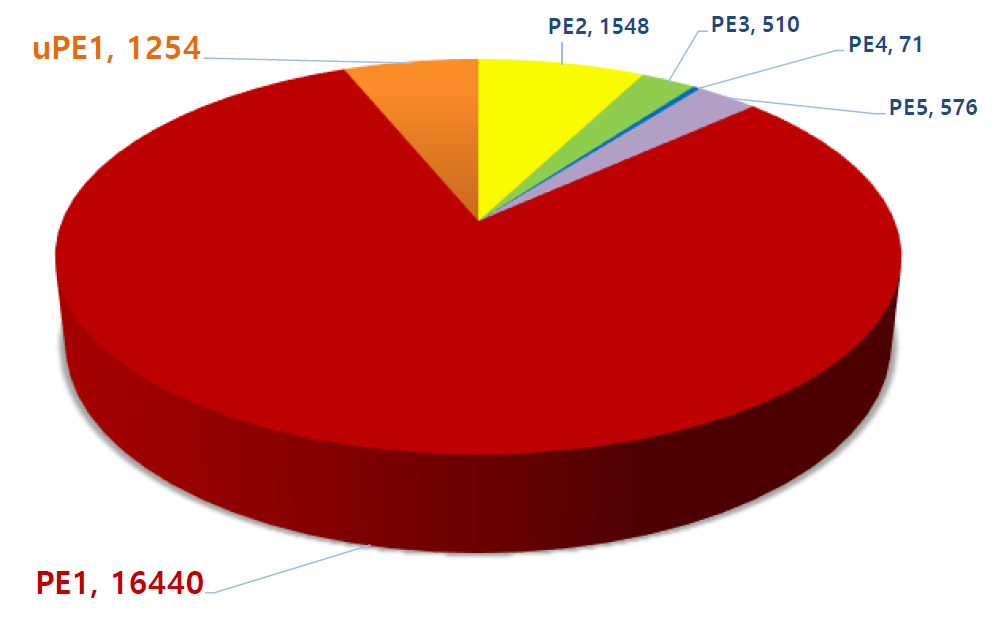
Update on the Missing Proteins and uPE1 (as of Jan. 11, 2019)
The initial goals of C-HPP are to identify at least one representative proteins encoded by each of the 21823 human genes (Ensembl, Apr 2011) and organize the data in accordance with the chromosomal structures. A second goal is to identify peptides for each protein which is observed in a wide range of samples and is suitable for preparation of reference peptides. A third goal is to perform tissue localization and quantitative studies using either mass spectrometric approach and/or antibody captured reagents. A fourth goal is to identify the set of isoforms coded from each human gene, such as alternative splicing and nsSNPs.
C-HPP Mission and Objectives
The mission of the C-HPP is to map and annotate the entire human proteome comprising the individual proteins encoded by each chromosome, their major splice forms, mature N- and C-termini, and their major protein post-translational modifications (PTMs) (see HUPO.org). In the C-HPP this is accomplished by directed studies initiated by the 25-international chromosome + mitochondrial DNA teams. Effective collaborations exist between the chromosome teams and other members of HUPO within the 19 B/D-HPP initiatives and the 4 HPP Pillars.
Phase 1 of the HPP project is focused on identifying by mass spectrometry all human proteins, presently estimated in the human genome to be 20,399 (neXtProt 2019-01-11). Those proteins identified by protein existence (PE) information number some 17,694 (PE1), with PE2 – 4 proteins remaining to be detected at the protein level—the so called “missing proteins” (MPs). At present, there remain 2,129 MPs (PE 2 – 4) yet to be identified. In Santiago C-HPP-2018, the neXt-CP50 Challenge was launched to functionalize proteins in the “Dark Proteome” with no known function, whether predicted or described. In 2018 PE1 – 4 proteins these numbered 1,937.
Phase 2 will focus on the neXt-CP2000 (to functionalize 2,000 uPE1s), ~5 PTMs / PE1 protein, and their splice forms. Also targeted are nonconventional-encoded small open reading frame translation products (smORFs), fusion proteoforms, and translatable products of long non-coding (lnc) RNAs.
A typical experimental workflow of missing protein identification by C-HPP teams
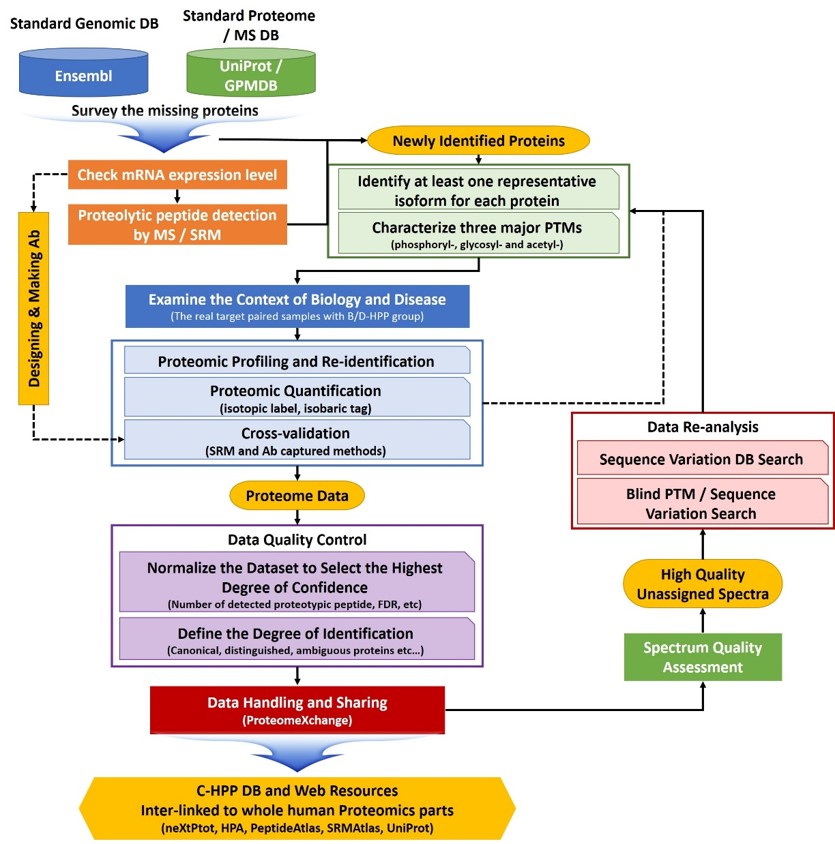
Stage 1. Experimental procedures for data production (Steps 1-5)
- Step 1 is to make a list of “missing proteins” using the several DBs (e.g., UniProt, Ensembl, GPMDB) by cross checking with an entire list of protein coding genes. At the same time, another effort will be made to improve the quality of mass spectrometric identifications in all but the highest probability category
- Step 2 will be to obtain specific mRNA expression pattern by RNA-seq and reverse transcription-polymerase chain reaction, based on public databases (GeneCards [www.genecards.org] and dbEST [www.ncbi.nlm.nig.gov/dbEST]) with defined expression thresholds. For these transcriptomic analyses, we will work with a group of cell biologists who have various specific cell lines and stem cells which may provide very unusual, rarely expressed proteins that are hard to detect under normal culture conditions
- Step 3 will be to characterize at least one representative isoform and three major translational modifications (PTMs) (i.e., phosphoryl-, glycosyl-, and acetyl-) for each protein.
- Step 4 will be to explore the annotation and disease related context of newly identified proteins in collaboration with the B/D project.
- Step 5 will be to perform the validation works including proteomic profiling with re-identification, quantifications and cross-validation (SRM and Ab captured methods).
Stage 2: Quality control of data and submission procedures (Steps 6-8)
- Step 6 will be to normalize the dataset to select the highest degree of confidence. Given the presence of two types of proteotypic peptides and ambiguously mapped peptides (www.proteomecenter.org), a more sensitive unambiguous, and quantitative assay should be developed for a protein. We will use SRMs as main engines for quantitation since the SRMAtlas is already available and provides suggested transitions and collision energy settings, observed retention times, calculated hydrophobicity, and information concerning peptide fragments.
- Step 7 will be to set the standard operational procedures for data handling. As we learned from the exploratory phase of the Human Plasma Proteome Project to consistently characterize proteins by MS methods, we should also put significant efforts into quality control and ensure commitment to deep analysis of specimens.
- Step 8 will be to build the C-HPP databases and utilize the data for biology and disease research in collaboration with B/D-HPP group. Findings and results generated by an individual group will belong to the corresponding investigators for publication. However, the results from each group should be published or deposited in the central C-HPP databases. It is also necessary to encourage all PIs to offer an opportunity of contribution and even re-analysis to the B/D and C-HPP teams.




